化学反硝化法脱除地下水中的硝酸盐
范彬,黄霞
(清华大学环境科学与工程系,北京100084)
摘 要:综述了地下水中的硝酸盐采用化学反硝化方法进行处理的最新研究进展。与生物反硝化法相比,化学反硝化法具有潜在的经济优势和对小型及分散给水处理的适应性。以铁为代表的活泼金属还原法的特点是还原剂价格低廉、来源广泛,但反硝化的产物主要是氨氮。化学催化反硝化方法被认为是最有应用前景的脱除地下水中硝酸盐的方法,但受反应过程中传质因素的限制,目前在催化活性和选择性方面还不够理想。
关键词:地下水;硝酸盐;化学反硝化;催化反硝化
中图分类号:TU991.11+2
文献标识码:B
文章编号:1000-4602(2001)11-0027-05
人类大规模的固氮活动(如工业固氮和大规模生物固氮)破坏了自然界氮素循环的平衡,很久以来地球上每年的固氮量远远超过通过反硝化释放的氮素量,从而导致硝酸盐氮在地下水中的积累,并日趋严重。摄入过多的硝酸盐对人的健康有多方面的危害,如引起高铁血蛋白症及诱发癌症等,对牲畜也有多方面的危害。世界卫生组织早就规定饮用水中的硝酸盐氮<10mg/L,而我国的标准仍为20mg/L。
脱除地下水中硝酸盐污染的方法有多种,目前已经投入实际应用的主要是生物反硝化、离子交换及反渗透工艺[1]。尽管一些物化方法可以将硝酸盐从饮用水中分离出来,但解决地下水中硝酸盐污染的根本出路还是要恢复自然界氮素循环的平衡:①限制人工固氮并倡导清洁的农业生产;②采取人工方式促进氮素以氮气形式释放入大气。将水中硝酸盐氮还原为氮气的方法可以分为两大类:生物反硝化和化学反硝化。生物反硝化法早在20世纪80年代初就已实际应用,但仍存在一些缺点:①工艺复杂、运行管理要求高;②给被处理水造成各种不同程度的二次污染,一般需要后续处理;③反硝化速度慢、所需反应器体积庞大、建设费用高;④不太适合用于小规模及分散给水处理。与生物反硝化相比,化学反硝化法有两个潜在的突出优点:①单位体积反应器的脱硝速度可以比生物反硝化法快得多;②工艺简单,对运行管理的要求低。
1 化学反硝化法
就化学还原法而言,无论采取何种还原剂(能)或还原方式,基本上可以肯定硝酸盐氮首先被还原为亚硝酸盐氮,继而被还原为氮气或氨氮。从亚硝酸盐氮继续还原可能要经过NO或N2O阶段,但目前对硝酸盐氮的还原反应历程缺乏一致的认识。
迄今为止对化学反硝化法脱除饮用水中硝酸盐氮所进行的研究仅局限于采用氢气、活泼金属以及甲酸、甲醇等数种还原剂。现将其分为活泼金属还原法与催化反硝化法两大类。前者是以铁、铝、锌等金属单质为还原剂,后者指以氢气以及甲酸、甲醇等为还原剂,一般都必须有催化剂存在才能使反应进行。
2 活泼金属还原法
一些活泼金属,如Cd、Cd-Hg齐、铝、Devarda合金、锌、Arndt合金等,在碱性环境中可以使硝酸盐还原为亚硝酸盐或氨氮。
2.1 铝还原法
Murphy A.P.[2]研究了铝粉对饮用水中硝酸盐氮的还原作用。pH值对其影响很大,并且水中的硫酸盐硫也有可能同时被还原。当pH<8时,投入铝粉不会使硝酸盐氮和硫酸盐硫还原;而当pH>11.5时,水中同时发生硝酸盐氮和硫酸盐硫的还原反应;只有在9<pH<10.5时,才可以优先还原硝酸盐氮。反应的主要产物为氨氮(60%~95%),其次为亚硝酸盐氮和氮气,反应在十几分钟内就可以完成。这一工艺有两个主要缺点:一是需要精确控制体系的pH值,否则易于发生铝粉的钝化,并因与水或硫酸盐的反应增加铝粉用量;二是主要的反硝化产物是氨氮和亚硝酸盐氮,它们在饮用水中的允许浓度都比硝酸盐氮低得多,因而必须在后续处理中加以脱除。
2.2 铁还原法
铁还原法是被研究得最多的课题。Yong G.K.等人[3]在1964年就尝试以铁粉作还原剂脱除饮用水中硝酸盐氮,由于约75%的硝酸盐氮转化为氨氮,并且反应中需要调节pH值,作者认为这一方法在大规模饮用水处理中应用的前景有限。
20世纪90年代后,以铁为还原剂脱除饮用水中硝酸盐氮的方法重新受到关注。铁粉可与硝酸根发生快速反应,主要产物为氮气和亚硝酸根,总的反应可用下式表示[4]:
10Fe+6NO3-+3H2O→5Fe2O3+6OH-+3N2
Fe+NO3-+2H+→Fe3++H2O+NO2-
Cheng F.[5]等(1997年)研究了在有氧条件和不同pH值下的325目铁粉对硝酸盐氮的还原作用。反应是一个放热的过程(460 kJ/mol),反应产物为氨氮和Fe2+:
NO3-+10H++4Fe=NH4++3H2O+4Fe2+
由于反应中需要消耗氢离子,因而必须在反应过程中加入酸和采取pH的缓冲措施,否则该反应不能进行下去。反应速度快慢的依次是:pH=5>pH=6>pH=7。
铁粉还原法的原理都比较相似,故而有着共同的缺点:①反应中需要比较严格地调节pH值;②反应后的水中有较多的氨氮和硝酸盐氮,需要后续处理。
由于:①铁的来源广泛、价格低廉;②反应速度较快;③除了pH值外,对环境(如温度)并没有特别要求。铁还原法还会继续受到关注,目前出现了几种改进措施。Chew C.F.等[6](1998年)提出了一种利用铁为还原剂的反硝化方法,且利用了外加电场产生的电渗和电动作用驱动土壤中水流和硝酸根向铁源迁移。Choe S[7].提出了一种纳米铁粉反硝化法,反应须在无氧条件下进行。纳米铁粉(1~100nm)可以将硝酸盐氮还原为氮气,反应后的水中几乎没有其他中间产物和氨氮。在室温条件下反应迅速、脱硝完全,且无需调节pH值。如果纳米铁粉的成本不太高,这一方法应该有相当的应用前景。
2.3其他方法
除铝和铁的还原法外,有一项日本专利[8]提出了以锌为还原剂脱除废水中硝酸盐氮的方法,主要的反应产物也是氨氮、亚硝酸盐氮和氮气。若在一种未加以说明的催化剂的作用下,反应过程产生的亚硝酸盐氮将与氨氮相互作用,使最终反应产物以氮气为主。
活泼金属还原法的主要缺点在于反应的产物难以无害的氮气为主,并且会产生金属离子、金属氧化物或水合金属氧化物等的二次污染,因而对后处理要求较高,同时也使这类方法的应用前景受到影响,而应用于污水脱硝可能更为合适。
3 化学催化反硝化法
化学催化反硝化研究始于20世纪80年代末,德国学者Vorlop K.D.等人[9]最早提出以通入的氢气为还原剂,在负载型的二元金属催化剂(如PdCu-Al2O3)的作用下,将硝酸盐氮还原。
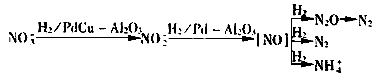
在理论上,只要合理选用催化剂和控制反应条件,通过化学催化反硝化完全可以将硝酸盐氮全部还原为氮气。化学催化剂的催化活性可以比生物反硝化酶的活性高30倍以上。由于上述过程可以在地下水的水质和水温条件下进行,若以氢气为还原剂还不会对被处理水产生二次污染,因此这一工艺原理受到密切关注,并被认为是最有发展前景的饮用水脱硝工艺。
催化剂多为贵金属,如Pd,Pt,Ru,Ir,Rh等。研究表明,Pd是将硝酸盐氮还原为氮气的最佳催化剂[10]。钯对NO3-→NO2-反应的催化效果并不理想,如在钯的表面掺以铜、锡、铟(辅助催化剂)等金属,则催化效果将大有改善,但这种二元合金催化剂对自NO2-以后的各反应步骤均无此作用。
催化反硝化是一个异相催化过程,唯有位于表面的金属原子才具备催化活性,因此应设法增加催化剂的比表面积。通常将活性金属以很薄的一层(几十nm)负载于惰性物质上,如氧化铝、氧化硅、沸石等,并制成一定形状的颗粒。这样既增加了活性金属的比表面,又使得在反应后易于实现催化剂与出水的分离。研究表明,催化剂载体的性质(如酸碱性)对催化反硝化过程有一定影响[11]。
影响水中催化反硝化反应的因素有很多,其中既有热力学方面的,更有动力学方面的。一些反应中间过程,诸如所有反应物、中间产物或最终产物向(或离开)催化剂表面的传质过程、吸附与解吸的过程、表面迁移与反应的过程,都有可能成为反应速度的控制步骤,从而影响脱除硝酸盐的速度和最终反应产物的组成,即影响催化活性和选择性。催化活性(activity)可以用单位质量催化剂在单位时间内脱除硝酸盐氮的量来表示;催化选择性(selectivity)可以用某一产物的产率或产量表示,通常指产物为氮气时的催化选择性。目前催化反硝化的选择性可以达到90%以上,这样的选择性还不能保证出水中的氨氮或亚硝酸盐氮的浓度满足饮用水的要求。
3.1 催化剂
催化剂的性质对催化反硝化反应的速度以及最终产物的组成有至关重要的影响。在催化剂的设计及制备方面虽然有一些理论指导,但经验的成分仍很多。组成相同的催化剂可能会因制备过程的细微差别而表现出不同的催化性质。目前一致认为,以金属钯为主的二元催化剂最适合充当硝酸盐氮还原的催化剂,而钯的一元催化剂最适合用来催化亚硝酸盐氮的还原反应。
辅助催化剂不仅影响催化剂的活性,而且影响催化剂的选择性[12](如表1)。
同时,不同方法制备的催化剂的最佳配比、活性与选择性也有比较大的差别[13](如表2)。
注:t=10 ℃,CO(NO-3)=2 mmol/L,pH=5,PH2=113 kPa。
3.2水质因素
虽然化学催化反硝化可以在地下水的水温、水质条件下进行,但地下水水质组成对二元(Pd-Cu)催化反硝化的活性和选择性都有一定的影响。一些研究表明,与用去离子水和硝酸盐配成的水相比,处理实际地下水时的催化反硝化活性和选择性都有不同程度的下降。水质因素的影响表现在以下几个方面:a.水中阳离子的作用影响硝酸根和氢氧根在水中的迁移速度;b.在催化剂表面形成沉淀或污垢,从而降低催化剂的有效表面;c.水中的阴离子与硝酸根竞争表面吸附或活性位置;d.水中可能出现的S2-等使催化剂中毒。
与生物反硝化一样,催化反硝化对水中的溶解氧也比较敏感。一方面,氧通过与硝酸盐氮争夺电子而抑制硝酸盐氮的还原;另一方面,水中的溶解氧还可能导致铜等辅助金属催化剂被氧化而失活。因此,在化学反硝化前必须将被处理水中的溶解氧脱除。
Pintar A等(1998年)研究了水中的永久硬度和盐效应对催化反硝化的影响。研究表明,不同金属硝酸盐的反硝化速度不同,硝酸盐氮在催化剂表面还原反应的表观速度常数随阳离子的离子势的增加而增加:K+<Na+<Ca2+<Mg2+<Al3+。水中的永久硬度对硝酸盐氮的还原活性与选择性都没有明显影响,但重碳酸根对硝酸盐氮的还原有很大影响,这可能是由于重碳酸根与硝酸根结构相似,二者在催化剂表面发生竞争吸附所致[14]。
水中的化学反硝化过程还有可能受到反应器中滋生的微生物影响[15],这种情形在反应器的长期运行中无法避免。这些微生物大多属于兼性厌氧菌,其中的一些自养菌可以氢气为电子供体进行生物反硝化,但可能仅进行到亚硝酸盐氮,目前对微生物的复杂影响的认识还很少。
3.3 pH值和传质过程
根据反应的电中性原则,在反硝化反应过程中任何消失的阴离子(NO-3或NO-2)或增加的阳离子(NH+4)都需要由反应中产生的OH-来平衡:
NO-3+H2=NO-2+H2O
2NO-2+4H2=N2+3H2O+2OH-2NO-2+6H2=2NH+4+4OH-
亦即在反硝化过程中催化剂与溶液界面处的pH值有升高的趋势。
pH值对催化反硝化的反应历程有很大的影响。随pH值的升高,无论是以二元催化剂催化还原硝酸盐氮,还是以一元催化剂催化还原亚硝酸盐氮,催化活性都要大幅下降,而氨氮的产率大幅上升。因此,必须采取一定措施来控制反应器内的pH值。另外,由于异相催化反硝化反应是一个表面的过程,为了保证某一表面活性点处的催化活性和选择性,必须能在该点处保持良好的pH值。也就是说,一方面应使表面反应产生的OH-尽快“移走”,另一方面应采取措施使溶液内部的H+尽快“移入”。因此,在催化反硝化过程中,除需控制一定的酸度外,还必须使催化剂的所有活性表面与溶液内部保持良好的传质效果。
目前大部分工艺使用的负载型催化剂的颗粒不仅有外部表面,而且有内部表面。显然,颗粒内表面与溶液内部的传质必定会面临一些困难,对于采用小颗粒催化剂的小型试验系统而言,严格控制催化剂绝大多数表面微环境中的pH值并不难,通常通过补充酸性物质(如二氧化碳或硫酸、盐酸等)及强烈的搅拌即可达到,然而在中试或生产性系统中,这一问题将会非常棘手。在更大型的系统中,较大粒径的负载型催化剂颗粒的内表面积在所有表面积中占有更高的比例,加上要在大型反应器内达到完全混合均匀并不是件容易的事,因此传质问题会变得更加严重,这是制约催化反硝化工艺实用化的最关键的难题。
目前在改善传质方面有几种措施。一种将微粒催化剂和胶体态催化剂填入中空纤维膜的工艺可以获得很高的催化选择性[16]。还有一种将催化剂微粒和胶体包埋于聚乙烯醇凝胶颗粒中的方法也可以获得较好的催化活性和选择性。据报道这种凝胶颗粒内的扩散传质性能非常好[17]。
膜催化剂[18~21]在催化反硝化中的应用是利用了膜的两种性质:a.对催化剂活性组分的固定作用;b.膜对参与反应的某一相的选择通过作用。可以实现氢气和被处理水从膜的两边进入并在催化剂的活性位置产生接触,从而改善传质效果,但其目前在催化选择性和活性两方面的效果还都不甚理想。现又有人提出纳米电极催化反硝化法,即将金属或合金催化剂制成纳米颗粒,并使之分散于被处理水中。采用微粒子电极方法,使纳米金属颗粒成为微粒子阴极,通过微电解过程在其表面产生氢,并通过其表面所具备的催化能力,使硝酸盐氮还原为氮气。利用膜技术实现纳米催化剂与出水分离,并利用碳材料为阳极,通过阳极氧化过程产生的二氧化碳为反硝化反应就地补充酸度,并保证反应器内的还原性环境。这一方法有以下几个主要优点:a.这种纳米金属颗粒不仅具有巨大的比表面积,而且所有的表面都是外表面,不存在负载型催化剂所面临的内部传质问题,可以同时获得高催化活性和选择性;b.纳米金属颗粒表面通过微电解产生的氢在产生之初是以原子形式吸附在表面活性位置,因此可以直接用于还原硝酸盐氮, 而无需像外加的氢气那样需要经过溶解—传质—吸附—解离成原子等一系列的过程;c.无需外加pH缓冲剂,反应器内的自我pH缓冲能力以及良好的传质性能可以使反硝化反应在几乎理 想的pH环境中进行;d.克服了外加氢气法所面临的氢气溶解度低(一般需加压溶解)、利用率 低以及运输和贮存要求高的缺点。这种仅需输入电能的工作模式特别适用于小型和分散给水的处理。
4 结论
在彻底消除地下水中硝酸盐污染和降低脱硝成本的两个方面,生物反硝化方法都是目前已投入实用的最好的方法。但生物方法仍有两个难以克服的缺点:①经济性仍不能令人满意;②管理要求较高,不适合用于小型或分散给水处理。与生物反硝化方法相比,化学反硝化方法反应速度快,对操作管理的要求低,具有潜在的经济性和对小型或分散给水处理的适应性。其中,以铁还原法为代表的活泼金属还原法由于还原剂价格低廉、反应速度快等原因而受到一些研究者的关注。但一般而言,这类方法反硝化的主要产物为氨氮,并且需要比较严格地控制pH值,因此只适用于污水脱硝。化学催化反硝化被一些学者认为是最有前景的地下水脱硝方法。与活泼金属还原法相比,化学催化反硝化方法可以将绝大部分硝酸盐氮转化为氮气(理论上可以使硝酸盐氮完全转化为氮气),且可以在地下水的水温下进行,并且催化活性比生物反硝化酶的活性高得多。尽管可以采用其他的一些还原剂,但以氢气为还原剂仍是研究的主要方向。目前,化学催化反硝化离实用化还有相当距离,存在的主要问题是反应过程中的传质因素影响了催化反硝化的活性和选择性,这将成为现阶段催化反硝化工艺研究的重点。
参考文献:
[1] 范彬,曲久辉,刘锁祥,等.饮用水中硝酸盐的脱除[J].环境污染治理技术与设备,2000,1(3):44-47.
[2]Murphy A P.Chemical removal of nitrate from water[J].Nature,1991 ,350:223-229.
[3]Sorg T J.Treatment technology to meet the interim primary drinking water regulat ions for inorganics[J].J AWWA,1978,70(2):105-118.
[4]Siantar D P,et al.Treatment of 1,2-dibromo-3-chloropropane and nitrate -contaminated water with zero-valent iron or hydrogen/pa lladium catalysts[J].Wat Res,1996,30(10):2351-2358.
[5]Cheng F,et al.Reduction of nitrate to ammonia by zero-valentiron[J].Chemosphere,1997,35(11):2689-2696.
[6]Chew C F,Zhang T C.In-situ remediation of nitrate -contaminated ground water by electrokinetics/iron wall process[J].Wa t Sci Tech,1998,38(7):135-146.
[7]Choe S,et al.Kinetics of reductive denitrification by nanoscale zero- valent iron[J].Chemosphere,2000,41:1307-1314.
[8]Yuasa Y,et al.JP08 192 169[96,192,169]JPN.Kokai Tokyo Koho[P].1996-06-30.
[9]Vorlop K D,Tacke T.Erste schritte auf dem weg zur edelmetall-kataly sierten nitr-und nitritentfernung aus trinkwasser[J].Chem Ing Tec h,1989,61:836-845.
[10]Hoerold S,Tacke T,Vorlop K D.Catalytic removal of nitrate and nitrite from drink ing water-1.Screening for hydrogenation catalysts and influence of reaction cond itions on activity and selectivity[J].Environ Tech,1993,14:931-945.
[11]Deganello F,et al.Catalytic reduction of nitrates and nitretes in water solu tion on pumice-supported Pd-Cu catalysts[J].Appl Catal B:Environ, 2000,24:265-268.
[12]Daum J,Vorlop K D.Kinetic investigation of the catalytic nitrate reduction:Construction of the test reactor system[J].Chem Eng Tech,1999,22:19 9-203.
[13]Prusse U,et al.Improving the catalytic nitrate reduction[J].Catal Today,20 00,55:79-88.
[14]Pintar A,Setinc M,Levec J.Hardness and salt effects on catalytic hydrogen of aqu eous nitrate solutions[J].J Catal,1998,174(1):72-79.
[15]Lecloux A J.Chemical,biological and physical constrains in catal ytic reduction p rocesses for purification of drinking water[J].Catal Today,1999,53:23-28.
[16]Hanlein M,et al.Preparation of microscopic catalysts and colloids for cataly sts nitrate and nitrite reduction and their use in a hallow fibre dialyser loop reactor[J].Prep Catal(Ⅶ),1998,118:99-107.
[17]Prusse U,et al.Encapsulation of microscopic catalysts in polyvinyl alcohol h ydrogel beads[J].Prep Catal(Ⅶ),1998,118:137-146.
[18]Strukul G,et al.Use of palladium based catalysts in the hydrogenation of nit rates in drinking water:from powders to membranes[J].Catal Today,2000,55:139- 148.
[19]Ludtke K,et al.Nitrate removal of drinking water by means of catalytically a ctive membranes[J].J Membrane Sci,1998,151:3-12.
[20]Ilinitch O M,et al.Catalytic membrane in reduction of aqueous nitrates:opera tional principles and catalytic performance[J].Catal Today,2000,50:137-146.
[21]Daub K,et al.Studies on the use of catalytic membranes for reduction of nitr ate in drinking water[J].Chem Eng Sci,1999,54:1577-1586.
E-mail:fanbinsy@tsinghua.edu.cn
收稿日期:2001-07-24
论文搜索
月热点论文
论文投稿
很多时候您的文章总是无缘变成铅字。研究做到关键时,试验有了起色时,是不是想和同行探讨一下,工作中有了心得,您是不是很想与人分享,那么不要只是默默工作了,写下来吧!投稿时,请以附件形式发至 paper@h2o-china.com ,请注明论文投稿。一旦采用,我们会为您增加100枚金币。



